Exert from Metabolites – August 2021
MDPI Concept Paper
Can the Mitochondrial Metabolic Theory Explain Better the Origin and Management of Cancer than Can the Somatic Mutation Theory?
Thomas N. Seyfried 1,* and Christos Chinopoulos 2 Citation: Seyfried, T.N.;
Chinopoulos, C. Can the Mitochondrial Metabolic Theory Explain Better the Origin and Management of Cancer than Can the Somatic Mutation Theory
Abstract: A theory that can best explain the facts of a phenomenon is more likely to advance knowledge than a theory that is less able to explain the facts. Cancer is generally considered a genetic disease based on the somatic mutation theory (SMT) where mutations in proto-oncogenes and tumor suppressor genes cause dysregulated cell growth. Evidence is reviewed showing that the mitochondrial metabolic theory (MMT) can better account for the hallmarks of cancer than can the SMT. Proliferating cancer cells cannot survive or grow without carbons and nitrogen for the synthesis of metabolites and ATP (Adenosine Triphosphate). Glucose carbons are essential for metabolite synthesis through the glycolysis and pentose phosphate pathways while glutamine nitrogen and carbons are essential for the synthesis of nitrogen-containing metabolites and ATP through the glutaminolysis pathway. Glutamine-dependent mitochondrial substrate level phosphorylation becomes essential for ATP synthesis in cancer cells that over-express the glycolytic pyruvate kinase M2 isoform (PKM2), that have deficient OxPhos, and that can grow in either hypoxia (0.1% oxygen) or in cyanide. The simultaneous targeting of glucose and glutamine, while elevating levels of nonfermentable ketone bodies, offers a simple and parsimonious therapeutic strategy for managing most cancers. Keywords: mutation
Introduction: Cancer is a systemic disease involving multiple time- and space-dependent changes in the health status of cells and tissues that lead to malignant tumors [1,2]. Dysregulated cell growth, i.e., neoplasia, is the biological endpoint of the disease [3]. Tumor cell invasion into surrounding tissues and their spread (metastasis) to distant organs is the primary cause of morbidity and mortality of most cancer patients [4–7]. Data from the American Cancer Society showed that the number of people dying in the US from cancer in 2013 was 580,350, and in 2020 it was 606,520, an increase of 4.3% [8,9]. The US population increase over this same period was 4.5%, indicating no real progress in cancer management. Cancer is predicted to overtake heart disease as the leading cause of death in Western societies. Is the failure to reduce the cancer death rate due to an incorrect theory on the origin of the disease? 2. Scientific Theories A scientific theory is simply an attempt to explain the facts of nature. Reality is based on replicated facts, whereas interpretation of the facts is based on credible theories. The heliocentric theory of Copernicus, Galileo, and Keppler was able to explain better the Metabolites 2021, 11, 572. https://doi.org/10.3390/metabo11090572 https://www.mdpi.com/journal/metabolites Metabolites 2021, 11, 572 2 of 21 movements of celestial bodies than was the geocentric theory of Ptolemy. The germ theory of Louis Pasteur was able to explain better the origin of contagious diseases than was the miasma “bad air” theory of Hippocrates and Galen. The Darwin–Wallace theory of evolution by natural selection was able to explain better the origin of species than was the theory of special creation [10]. In none of these examples could a hybrid theory be envisioned. A theory that can best explain the facts of a phenomenon is more likely to advance knowledge than is a theory less able to explain the facts. The provocative question before us is whether the Mitochondrial Metabolic Theory (MMT) can explain better the origin and management of cancer than can the current Somatic Mutation Theory (SMT)
According to the SMT, cancer is a complex genetic disease that arises from inherited or random somatic mutations in proto-oncogenes or in tumor suppressor genes [11–13]. While many mutations have been found in various tumors, the so-called driver gene mutations are considered most responsible for causing the disease [11,14]. Although the SMT is the dominant scientific explanation for the origin of cancer, numerous inconsistencies have emerged that seriously challenge the credibility of this theory [15–17]. The major inconsistencies include: (1) The absence of gene mutations and chromosomal abnormalities in some cancers [17–21]. For example, Greenman et al. found no mutations following extensive sequencing in 73/210 cancers [13], whereas Parsons et al., found no mutations in the P53, the PI3K, or the RB1 pathways in the Br20P tissue sample of a glioblastoma patient [22]. Cancer cells with no mutations should not exist according to the SMT. (2) The identification and clonal expansion of numerous driver gene mutations in a broad range of normal human tissues [23–27]. If driver genes cause cancer according to the SMT, then how is it possible that so many driver genes are found in normal human tissues that do not express cancer? No explanation has been presented on how the SMT can account for, (a) malignant tumors that have no mutations, or (b) normal cells that express driver mutations, but do not develop tumors [14]. (3) The general absence of cancers in chimpanzees despite having about 98% gene and protein sequence identity with humans even at the BRCA1 locus [28–31]. Despite anatomical differences between the breasts of humans and chimpanzees, breast cancer has never been documented in a female chimpanzee [31]. As DNA replication would be similar in normal tissue stem cells in chimpanzees and humans, the rarity of cancer in all chimpanzee organs undermines the “bad luck” hypothesis of Tomasetti and Vogelstein that cancer risk is due to random mutations arising during DNA replication in normal, noncancerous stem cells [32]. The rarity of cancer in primitive humans and in chimpanzees suggests that environmental factors (diet and lifestyle), rather than genetic mutations, are largely responsible for cancer [31,33]. It is important to remember that nothing in either general biology or in cancer biology makes sense except in the light of evolution [34,35]. (4) Theodor Boveri, the person most recognized as the originator of the SMT [36,37], never directly studied cancer and was highly apologetic for his general lack of knowledge about the disease. Indeed, Boveri stated: “I have no personal experience worth mentioning in any of the numerous specialized fields of tumour research. My knowledge comes almost exclusively from books. Given this, it is inevitable that I am unaware of many reports in the literature, that I overestimate the significance of many known facts and that I do not set enough store by others. But this article will doubtless contain even more serious defects, as is so often the case when an author makes an incursion into a field with which he is unfamiliar” [38]. Most importantly, Boveri also mentioned that defects in the cytoplasm could just as well be responsible for cancer as defects in the nucleus. The distinguished British geneticist C. D. Darlington also emphasized the importance of the cytoplasm in the origin of cancer [39]. The most compelling evidence against the SMT comes from the nuclear/cytoplasm transfer experiments showing that growth-regulated cells can be produced from tumorigenic nuclei, as long as the tumorigenic nuclei are localized in the cytoplasm containing Metabolites 2021, 11, 572 3 of 21 normal mitochondria [40,41]
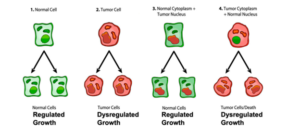
(Figure 1). Moreover, recent studies show that normal mitochondria can down-regulate multiple oncogenic pathways and abnormal growth in glioma, melanoma, and metastatic breast cancer cells [42–46]. These findings indicate that normal mitochondrial function can suppress dysregulated cell growth regardless of the number of gene or chromosomal abnormalities that might be present in the tumor nucleus. Although the somatic mutations present in the cancer nuclei of developing frogs and mice did not cause dysregulated cell growth, they did abort development suggesting an inhibitory lethal effect on the proliferation of normal cells [40]. If nuclear encoded driver genes were responsible for dysregulated cancer cell growth, then the results from the nuclear/mitochondrial transfer experiments would be opposite to the results shown in Figure 1. When viewed collectively, these findings imply that the nuclear somatic mutations found in many cancers cannot be the primary cause of the disease and seriously challenge the SMT as a credible explanation for the origin of cancer [14]. Despite these glaring inconsistencies, the SMT is presented as if it were a settled issue in most current college textbooks of genetics, biochemistry, and cell biology, as well as in the National Cancer Institute in stating that, “Cancer is a genetic disease—that is, it is caused by changes to genes that control the way our cells function, especially how they grow and divide” (12 October 2017) [40]. The view of cancer as a genetic disease has become a “silent assumption”, so completely accepted that it is no longer questioned. Could the continued acceptance of the SMT as an explanation for the origin of cancer be based more on dogmatic ideology than on rational thought [40,47]? If nuclear somatic mutations cannot be the origin of cancer, then how do cancer cells arise?
The Mitochondrial Metabolic Theory of Cancer
The Mitochondrial Metabolic Theory of Cancer According to the MMT, cancer arises from a gradual disruption of ATP synthesis through oxidative phosphorylation (OxPhos) leading to compensatory ATP synthesis through substrate level phosphorylation. It is defective OxPhos that ultimately causes most of the genomic changes in cancer, not the reverse. Although Otto Warburg is rightfully credited with the original discovery of cancer as a mitochondrial metabolic disease [50,51], he was unaware of information now available that more strongly supports the linkage between OxPhos deficiency and the origin of cancer (discussed later). The disruption of OxPhos leads to the accumulation of reactive oxygen species (ROS), which are mutagenic and carcinogenic [52–56]. The genomic instability and somatic mutations seen in most cancers arise as a consequence of the chronic production of ROS and acidification of the microenvironment [15,55,57–61]. In other words, the somatic mutations arise as downstream effects rather than as causes of cancer. The information summarized in Figure 1 shows that nuclear genomic mutations alone cannot be the origin of dysregulated cell growth, i.e., the signature phenotype of cancer. Could this information change opinions on the importance of mutations in the origin cancer? It is our view that the MMT can explain better the hallmarks and facts of cancer than can the SMT. The MMT is the only theory to provide a credible explanation for the “oncogenic paradox” that has perplexed the cancer field for decades [62–64]. Albert Szent-Gyorgyi first described the oncogenic paradox as a specific process (malignant transformation) that could be initiated by a plethora of unspecific events (radiation, asbestos, viral infections, rare inherited mutations, irritation, inflammation, chemicals etc.) [62]. Siddhartha Mukherjee also struggled to understand the paradox in stating on page 285 of his book: “What beyond abnormal, dysregulated cell division, was the common pathophysiological mechanism underlying cancer?” [64]. We solved the paradox in showing how the protracted loss of OxPhos, following mitochondrial damage, is the common pathophysiological mechanism responsible for the oncogenic paradox and the origin of cancer (Figure 2). .
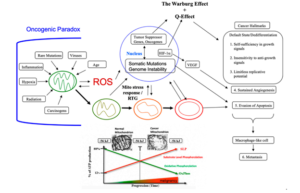
Figure 2. Cancer as a Mitochondrial Metabolic Disease. Cancer can arise from any number of unspecific influences (risk factors) that would alter the number, structure, and function of mitochondria thus affecting energy production through OxPhos. Unspecific cancer risk factors can include, age, viral infections, the Ras oncogene, rare inherited mutations, chronic inflammation, intermittent hypoxia, radiation exposure, chemical carcinogens etc. [2,65–68]. Any of these risk factors could cause chronic damage to OxPhos thus increasing the production of reactive oxygen species (ROS), which would ultimately Metabolites 2021, 11, 572 5 of 21 link to the six major hallmarks of cancer [2,12,68]. The process by which each of these unspecific risk factors can chronically damage OxPhos was described previously in detail [16,66,69,70]. Excessive ROS, mostly generated from OxPhos dysfunction, are carcinogenic and mutagenic and would cause significant damage to lipids, proteins, and nucleic acids in both the mitochondria and the in the nucleus [71]. Nuclear genomic instability, including the vast array of somatic mutations and aneuploidy, would arise because of ROS damage together with extracellular acidification and inflammation through a bidirectional interaction between the provocative agent and cells within a tissue [1,2,57,72,73]. Indeed, mutations in the p53 tumor suppressor gene and genomic instability have been linked directly to OxPhos deficiency and mitochondrial ROS production in cancer stem cells [55,74]. Fermentation metabolism and ROS formation underlie the hyperproliferation of tumor cells. A gradual reduction in OxPhos efficiency would elicit a mitochondrial stress response through retrograde (RTG) signaling [69,75–77]. RTG activation would cause persistent expression of various oncogenes, e.g., Hif-1a and c-Myc, that upregulate receptors and enzymes in both the glycolysis and the glutaminolysis pathways [75,78–82]. Oncogenes become facilitators of fermentation metabolism. ATP synthesis through mSLP (Q effect) will compensate for lost ATP synthesis through OxPhos or from PKM2 expression in glycolysis [83,84]. The path to carcinogenesis will occur only in those cells capable of sustaining energy production through substrate level phosphorylation, (SLP). Cells unable to replace OxPhos with SLP, e.g., CNS neurons or cardiomyocytes, would die and rarely become tumorigenic. Despite the shift from respiration to SLP, the ∆G’ATP hydrolysis remains fairly constant at approximately −56 kJ, indicating that the energy from SLP compensates for the reduced energy from OxPhos. When respiration becomes unable to maintain energy homeostasis, the RTG will initiate oncogene up-regulation and tumor suppressor gene inactivation. Protracted RTG activation becomes necessary to maintain the viability of incipient cancer cells. Genomic instability will arise as a secondary consequence of protracted mitochondrial stress from disturbances in the intracellular and extracellular environments. Metastasis arises from respiratory damage in cells of myeloid/macrophage origin either directly or after fusion hybridization with epithelialderived tumor cells [4,85]. Tumor progression and degree of malignancy is linked directly to ultrastructure abnormalities (mitochondrial cristolysis) and to the energy transition from OxPhos to substrate level phosphorylation (Warburg effect and Q effect) [83]. The T signifies an arbitrary threshold when the shift from OxPhos to SLP becomes irreversible. This scenario links all major cancer hallmarks to an extrachromosomal and epigenetic respiratory dysfunction and can explain the oncogenic paradox [70]. Reprinted with modifications from [68,83]. Since no inherited cancer mutation has been found that is 100% penetrant, inherited cancer mutations are also considered secondary effects and not primary causes of cancer [66,86–88]. If an inherited or somatic mutation were to be found in all cancers, it could be considered a primary cause of cancer. Such mutations, however, have not been found. Inherited cancer mutations could cause cancer if they compromise OxPhos function, making OxPhos dysfunction the primary cause of cancer. While mtDNA mutations have been found in many cancers [89], we were unable to find a single pathogenic mutation in the fully sequenced mtDNA of five independently-derived mouse brain tumors [90]. These findings indicate that mtDNA mutations alone cannot be the origin of all cancers. The mtDNA mutations are considered secondary risk factors and can be linked to cancer origin only if they also disrupt OxPhos function [91]. A chronic disruption of ATP synthesis through OxPhos would induce, by necessity, a compensatory energy production through the process of substrate level phosphorylation in both the cytoplasm and in the mitochondria. As normal mitochondrial function maintains cellular differentiation, the rewiring of energy metabolism from respiration to fermentation would cause dedifferentiation and dysregulated proliferation [2,40,50,51,58]. While aerobic fermentation (Warburg effect) is considered another emerging hallmark of cancer [12], the replacement of abnormal mitochondria with normal mitochondria will also reverse this hallmark [44,46,92]. In other words, OxPhos sufficiency will reverse the Warburg effect [44,93]. Hence, the energy transition from respiration to fermentation can explain the major hallmarks of cancer, as described by Hanahan and Weinberg (Figure 2). Metabolites 2021, 11, 572 6 of 21 A chronic loss of OxPhos will activate the mitochondrial stress response or the retrograde (RTG) signaling system [75–77]. Activation of this system stabilizes Hif-1α and upregulates the expression of c-Myc, key oncogenes necessary for the upregulation of substrate level phosphorylation through the glycolysis and the glutaminolysis pathways, respectively [83]. As plasma membrane pumps are perpetual consumers of ATP, no cell can survive for very long without constant synthesis of ATP for the pumps [94]. The transition from ATP synthesis through OxPhos to ATP synthesis through fermentation thus becomes essential for cell viability. Moreover, the energy transition from OxPhos to fermentation will cause a cell to enter its “default” state. Proliferation is the evolutionary conserved default state of metazoan cells, once freed from active control [3,17]. The mitochondrial OxPhos system provides the active control necessary for maintaining the quiescent or differentiated state. The protracted replacement of ATP synthesis through OxPhos with ATP synthesis through substrate level phosphorylation will cause the cell to enter its default state of proliferation [62]. Szent-Gyorgyi described how unbridled proliferation, driven by fermentation metabolism, was the common phenotype of all cells before oxygen entered the atmosphere some 2.5 billion years ago [62]. Based on the concepts of evolutionary biology, the transition from respiration to fermentation becomes the most logical explanation for the first three hallmarks of cancer involving dysregulated cell growth (Figure 2). The acidification of the cancer microenvironment, arising from the excretion of fermentation end products, e.g., lactate and succinate, will initiate angiogenesis. This process, however, can be bi-directional leading to an escalating situation of biological chaos [40,95]. Stabilization of Hif-1α is ultimately responsible for angiogenesis, i.e., the fourth hallmark [82,83,96,97]. As mitochondria control apoptosis [98], evasion of apoptosis would be an expected outcome of dysfunctional mitochondria and can account for the fifth cancer hallmark. While the rewiring of energy production from OxPhos to substrate level phosphorylation can easily explain the first five cancer hallmarks, how might this energy rewiring be linked to metastasis, the sixth major cancer hallmark? Emerging evidence indicates that metastasis involves transformation of myeloid cells or fusion hybridization between macrophages and transformed epithelial cells [4,99–107]. Macrophages and myeloid cells are mesenchymal cells that are already programed to migrate through tissues, to intravasate blood vessels, to function in the circulation, and to extravasate blood vessels for involvement in tissue repair and wound healing [4,108–110]. Similar to macrophages, many metastatic cancer cells are immunosuppressive and express phagocytic behavior [100,111–113]. The absence of metastasis in crown-gall plant cancers, despite expressing aerobic fermentation (Warburg effect), is due to the absence of a cellular immune system (macrophages and lymphocytes) in plants [4,7]. Macrophages can acquire mitochondria with dysfunctional OxPhos through various fusion hybridization events with neoplastic stem cells in an acidic and hypoxic microenvironment [101,114] (Figure 3). Radiation therapy can also facilitate tumor cell-macrophage/microglial fusion-hybridization thus producing highly invasive metastatic cells, as an unintended consequence [115,116]. It is also interesting that glutamine is a major energy metabolite for cells of the immune system including macrophages [117–119]. This fact could account in part for the glutamine dependency of metastatic cancer cells [120–123]. As macrophages are immunosuppressive, metastatic cells with macrophage properties would be powerful suppressors of the immune system. These properties could contribute to the failure of some immunotherapies [124,125]. The transition from respiration to fermentation can also explain the drug resistance of metastatic tumor cells [126,127]. The drug resistance of tumor cells is due in large part to the replacement of energy synthesis from OxPhos to fermentation [2]. Hence, the control of metastasis can be improved with better knowledge of macrophage biology. Figure 3.
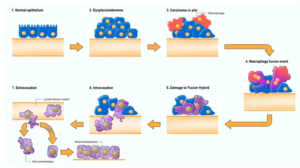
Fusion-hybrid hypothesis for cancer cell metastasis. According to the fusion hybrid hypothesis, metastatic cancer cells can arise following fusion-hybridization between neoplastic epithelial cells and myeloid cells (macrophages). The fusion hybrid hypothesis originated with the work of Aichel in 1911 and was expanded by the Pawelek and the Munzarova groups [105,128–132]. Macrophages are known to invade in situ carcinoma as if it were an unhealed wound [95,109,133]. This creates a protracted inflammatory microenvironment leading to fusion hybridization between the neoplastic epithelial cell and the mesenchymal macrophage. Mitochondrial damage becomes the driver for the neoplastic transformation of the epithelial cell and of the fusion hybrids. Inflammation damages mitochondria leading to enhanced fermentation and acidification of the microenvironment. The gradual replacement of normal macrophage mitochondria with dysfunctional mitochondria in the hybrid cell cytoplasm leads to rogue behavior in cells that naturally possess the capability to, (1) move through tissues, (2) suppress the immune system, (3) enter (intravasate), and to exit (extravasate) the circulation. In addition to explaining the “seed-soil” hypothesis of metastasis, the fusion hybrid hypothesis can also explain how metastatic cells can re-capitulate the epithelial characteristics of the primary tumor at secondary micro-metastatic growth sites [4,85]. Furthermore, this hypothesis can explain the phenomenon of mesenchymal epithelial transition without invoking a mutation suppression mechanism. See text for more details. Modified from [85,134]. The macrophage/myeloid origin of metastasis, based on the mitochondrial metabolic theory, should be compared with the epithelial mesenchymal transition (EMT) and mesenchymal epithelial transition (MET) for the origin of metastasis, based on the somatic mutation theory [4,12]. It is unclear how random somatic mutations could be responsible for metastasis, as the metastatic cascade is a non-random phenomenon that is common to many cancer types [5]. Each part of the cascade involves an ordered regulation of evolutionary conserved biological processes. Moreover, the metastatic behavior of cells can occur in the absence of mutations [7,135,136]. The EMT/MET hypothesis has yet to explain how random somatic mutations could transform an epithelial cell into a biologically distinct mesenchymal cell (EMT), and then have these random mutations be suppressed or reversed to allow a transition of the mesenchymal phenotype back to an epithelial phenotype (MET) [12,134]. We consider these fantastical biological transitions as inconsistent with evolutionary biology [85]. In summary, the mitochondrial metabolic theory can explain better the facts of metastasis than can the somatic mutation theory. Figure 3. Fusion-hybrid hypothesis for cancer cell metastasis. According to the fusion hybrid hypothesis, metastatic cancer cells can arise following fusion-hybridization between neoplastic epithelial cells and myeloid cells (macrophages). The fusion hybrid hypothesis originated with the work of Aichel in 1911 and was expanded by the Pawelek and the Munzarova groups [105,128–132]. Macrophages are known to invade in situ carcinoma as if it were an unhealed wound [95,109,133]. This creates a protracted inflammatory microenvironment leading to fusion hybridization between the neoplastic epithelial cell and the mesenchymal macrophage. Mitochondrial damage becomes the driver for the neoplastic transformation of the epithelial cell and of the fusion hybrids. Inflammation damages mitochondria leading to enhanced fermentation and acidification of the microenvironment. The gradual replacement of normal macrophage mitochondria with dysfunctional mitochondria in the hybrid cell cytoplasm leads to rogue behavior in cells that naturally possess the capability to, (1) move through tissues, (2) suppress the immune system, (3) enter (intravasate), and to exit (extravasate) the circulation. In addition to explaining the “seed-soil” hypothesis of metastasis, the fusion hybrid hypothesis can also explain how metastatic cells can re-capitulate the epithelial characteristics of the primary tumor at secondary micro-metastatic growth sites [4,85]. Furthermore, this hypothesis can explain the phenomenon of mesenchymal epithelial transition without invoking a mutation suppression mechanism. See text for more details. Modified from [85,134]. The macrophage/myeloid origin of metastasis, based on the mitochondrial metabolic theory, should be compared with the epithelial mesenchymal transition (EMT) and mesenchymal epithelial transition (MET) for the origin of metastasis, based on the somatic mutation theory [4,12]. It is unclear how random somatic mutations could be responsible for metastasis, as the metastatic cascade is a non-random phenomenon that is common to many cancer types [5]. Each part of the cascade involves an ordered regulation of evolutionary conserved biological processes. Moreover, the metastatic behavior of cells can occur in the absence of mutations [7,135,136]. The EMT/MET hypothesis has yet to explain how random somatic mutations could transform an epithelial cell into a biologically distinct mesenchymal cell (EMT), and then have these random mutations be suppressed or reversed to allow a transition of the mesenchymal phenotype back to an epithelial phenotype (MET) [12,134]. We consider these fantastical biological transitions as inconsistent with evolutionary biology [85]. In summary, the mitochondrial metabolic theory can explain better the facts of metastasis than can the somatic mutation theory.
Source: Metabolites 2021, 11, 572. https://doi.org/ 10.3390/metabo11090572 Academic Editor: Daniel Oscar Cicero Received: 11 July 2021 Accepted: 20 August 2021 Published: 25 August 2021 Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. Copyright: © 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https:// creativecommons.org/licenses/by/ 4.0/). 1 Department of Biology, Boston College, Chestnut Hill, MA 02467, USA 2 Department of Medical Biochemistry, Semmelweis University, 1094 Budapest, Hungary; chinopoulos.christos@med.semmelweis-univ.hu * Correspondence: Thomas.seyfried@bc.edu
References
- Seyfried, T.N.; Yu, G.; Maroon, J.C.; D’Agostino, D.P. Press-pulse: A novel therapeutic strategy for the metabolic management of cancer. Nutr. Metab. 2017, 14, 19. [CrossRef]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D’Agostino, D.P. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis 2014, 35, 515–527. [CrossRef] [PubMed]
- Sonnenschein, C.; Soto, A.M. Somatic mutation theory of carcinogenesis: Why it should be dropped and replaced. Mol. Carcinog. 2000, 29, 205–211. [CrossRef]
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [CrossRef] [PubMed]
- Fidler, I.J. The pathogenesis of cancer metastasis: The ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [CrossRef]
- Lazebnik, Y. What are the hallmarks of cancer? Nat. Rev. Cancer 2010, 10, 232–233. [CrossRef]
- Tarin, D. Cell and tissue interactions in carcinogenesis and metastasis and their clinical significance. Semin. Cancer Biol. 2011, 21, 72–82. [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [CrossRef]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [CrossRef]
- Darwin, C. On the Origin of Species by Means of Natural Selection, or on the Preservation of Favored Races in the Struggle for Life; John Murry: London, UK, 1859; 513p.
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [CrossRef]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [CrossRef]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. [CrossRef] [PubMed]
- Seyfried, T.N.; Mukherjee, P.; Iyikesici, M.S.; Slocum, A.; Kalamian, M.; Spinosa, J.P.; Chinopoulos, C. Consideration of Ketogenic Metabolic Therapy as a Complementary or Alternative Approach for Managing Breast Cancer. Front. Nutr. 2020, 7, 21. [CrossRef]
- Seyfried, T.N. Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer; John Wiley & Sons: Hoboken, NJ, USA, 2012; 421p.
- Soto, A.M.; Sonnenschein, C. The somatic mutation theory of cancer: Growing problems with the paradigm? Bioessays 2004, 26, 1097–1107. [CrossRef] [PubMed]
- Bayreuther, K. Chromosomes in primary neoplastic growth. Nature 1960, 186, 6–9. [CrossRef] [PubMed]
- Pitot, H.C. Some biochemical aspects of malignancy. Ann. Rev. Biochem. 1966, 35, 335–368. [CrossRef]
- Baker, S.G. A cancer theory kerfuffle can lead to new lines of research. J. Natl. Cancer Inst. 2015, 107, dju405. [CrossRef]
- Braun, A.C. On the origin of the cancer cells. Am. Sci. 1970, 58, 307–320. 22. Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [CrossRef]
- Yizhak, K.; Aguet, F.; Kim, J.; Hess, J.M.; Kübler, K.; Grimsby, J.; Frazer, R.; Zhang, H.; Haradhvala, N.J.; Rosebrock, D.; et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 2019, 364, eaaw0726. [CrossRef]
- Yokoyama, A.; Kakiuchi, N.; Yoshizato, T.; Nannya, Y.; Suzuki, H.; Takeuchi, Y.; Shiozawa, Y.; Sato, Y.; Aoki, K.; Kim, S.K.; et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 2019, 565, 312–317. [CrossRef]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [CrossRef] 26. Martincorena, I.; Fowler, J.C.; Wabik, A.; Lawson, A.R.; Abascal, F.; Hall, M.W.; Cagan, A.; Murai, K.; Mahbubani, K.; Stratton, M.R.; et al. Somatic mutant clones colonize the human esophagus with age. Science 2018, 362, 911–917. [CrossRef]
- Chanock, S.J. The paradox of mutations and cancer. Science 2018, 362, 893–894. [CrossRef] [PubMed]
- Lowenstine, L.J.; McManamon, R.; Terio, K.A. Comparative Pathology of Aging Great Apes: Bonobos, Chimpanzees, Gorillas, and Orangutans. Vet. Pathol. 2016, 53, 250–276. [CrossRef] [PubMed]
- Huttley, G.A.; Easteal, S.; Southey, M.C.; Tesoriero, A.; Giles, G.G.; McCredie, M.R.; Hopper, J.L.; Venter, D.J. Adaptive evolution of the tumour suppressor BRCA1 in humans and chimpanzees. Australian Breast Cancer Family Study. Nat. Genet. 2000, 25, 410–413. [CrossRef]
Metabolites 2021, 11, 572 15 of 21 30. Puente, X.S.; Velasco, G.; Gutierrez-Fernandez, A.; Bertranpetit, J.; King, M.C.; Lopez-Otin, C. Comparative analysis of cancer genes in the human and chimpanzee genomes. BMC Genom. 2006, 7, 15. [CrossRef] [PubMed]
- Varki, N.M.; Varki, A. On the apparent rarity of epithelial cancers in captive chimpanzees. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140225. [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [CrossRef]
- Kopp, W. How Western Diet and Lifestyle Drive the Pandemic of Obesity and Civilization Diseases. Diabetes Metab. Syndr. Obes. 2019, 12, 2221–2236. [CrossRef] [PubMed]
- Dobzhansky, T. Nothing in biology makes sense except in the light of evolution. Am. Biol. Teach. 1973, 35, 125–129. [CrossRef]
- Seyfried, T.N. Nothing in cancer biology makes sense except in the light of evolution. Chapter 15. In Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer; John Wiley & Sons: Hoboken, NJ, USA, 2012; pp. 261–275.
- Barrett, J.C. Mechanisms of multistep carcinogenesis and carcinogen risk assessment. Environ. Health Perspect. 1993, 100, 9–20. [CrossRef]
- Knudson, A.G. Cancer genetics. Am. J. Med. Genet. 2002, 111, 96–102. [CrossRef]
- Boveri, T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J. Cell Sci. 2008, 121 (Suppl. S1), 1–84. [CrossRef]
- Darlington, C.D. The plasmagene theory of the origin of cancer. Br. J. Cancer 1948, 2, 118–126. [CrossRef]
- Seyfried, T.N. Cancer as a mitochondrial metabolic disease. Front. Cell Dev. Biol. 2015, 3, 43. [CrossRef] [PubMed]
- Seyfried, T.N. Mitochondria: The ultimate tumor suppressor. Chapter 11. In Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer; John Wiley & Sons: Hoboken, NJ, USA, 2012; pp. 195–205.
- Kaipparettu, B.A.; Ma, Y.; Park, J.H.; Lee, T.L.; Zhang, Y.; Yotnda, P.; Creighton, C.J.; Chan, W.Y.; Wong, L.J. Crosstalk from non-cancerous mitochondria can inhibit tumor properties of metastatic cells by suppressing oncogenic pathways. PLoS ONE 2013, 8, e61747. [CrossRef]
- Chang, J.C.; Chang, H.S.; Wu, Y.C.; Cheng, W.L.; Lin, T.T.; Chang, H.J.; Kuo, S.J.; Chen, S.T.; Liu, C.S. Mitochondrial transplantation regulates antitumour activity, chemoresistance and mitochondrial dynamics in breast cancer. J. Exp. Clin. Cancer Res. 2019, 38, 30. [CrossRef]
- Fu, A.; Hou, Y.; Yu, Z.; Zhao, Z.; Liu, Z. Healthy mitochondria inhibit the metastatic melanoma in lungs. Int. J. Biol. Sci. 2019, 15, 2707–2718. [CrossRef]
- Ma, Y.; Bai, R.K.; Trieu, R.; Wong, L.J. Mitochondrial dysfunction in human breast cancer cells and their transmitochondrial cybrids. Biochim. Biophys. Acta 2010, 1797, 29–37. [CrossRef]
- Sun, C.; Liu, X.; Wang, B.; Wang, Z.; Liu, Y.; Di, C.; Si, J.; Li, H.; Wu, Q.; Xu, D.; et al. Endocytosis-mediated mitochondrial transplantation: Transferring normal human astrocytic mitochondria into glioma cells rescues aerobic respiration and enhances radiosensitivity. Theranostics 2019, 9, 3595–3607. [CrossRef]
- Mayr, E. The Growth of Biological Thought: Diversity, Evolution, and Inheritance; Belknap Harvard: Cambridge, MA, USA, 1982; 974p.
- Goodman, S.N.; Fanelli, D.; Ioannidis, J.P. What does research reproducibility mean? Sci. Transl. Med. 2016, 8, 341ps12. [CrossRef] [PubMed]
- McNutt, M. Journals unite for reproducibility. Science 2014, 346, 679. [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [CrossRef] [PubMed]
- Warburg, O. On the respiratory impairment in cancer cells. Science 1956, 124, 269–270. [PubMed]
- Newman, D.L.; Gregory, S.L. Co-Operation between Aneuploidy and Metabolic Changes in Driving Tumorigenesis. Int. J. Mol. Sci. 2019, 20, 4611. [CrossRef]
- Valle, A.; Oliver, J.; Roca, P. Role of uncoupling proteins in cancer. Cancers 2010, 2, 567–591. [CrossRef]
- Hammond, E.M.; Dorie, M.J.; Giaccia, A.J. ATR/ATM targets are phosphorylated by ATR in response to hypoxia and ATM in response to reoxygenation. J. Biol. Chem. 2003, 278, 12207–12213. [CrossRef]
- Bartesaghi, S.; Graziano, V.; Galavotti, S.; Henriquez, N.V.; Betts, J.; Saxena, J.; Minieri, V.; Deli, A.; Karlsson, A.; Martins, L.M.; et al. Inhibition of oxidative metabolism leads to p53 genetic inactivation and transformation in neural stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, 1059–1064. [CrossRef
- Klaunig, J.E.; Kamendulis, L.M.; Hocevar, B.A. Oxidative stress and oxidative damage in carcinogenesis. Toxicol. Pathol. 2010, 38, 96–109. [CrossRef] [PubMed]
- Sonugur, F.G.; Akbulut, H. The Role of Tumor Microenvironment in Genomic Instability of Malignant Tumors. Front. Genet. 2019, 10, 1063. [CrossRef] [PubMed]
- Fosslien, E. Cancer morphogenesis: Role of mitochondrial failure. Ann. Clin. Lab. Sci. 2008, 38, 307–329. [PubMed] 59. Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [CrossRef] [PubMed]
- Desler, C.; Lykke, A.; Rasmussen, L.J. The effect of mitochondrial dysfunction on cytosolic nucleotide metabolism. J. Nucleic Acids 2010, 2010, 701518. [CrossRef] [PubMed]
- Degtyareva, N.P.; Heyburn, L.; Sterling, J.; Resnick, M.A.; Gordenin, D.A.; Doetsch, P.W. Oxidative stress-induced mutagenesis in single-strand DNA occurs primarily at cytosines and is DNA polymerase zeta-dependent only for adenines and guanines. Nucleic Acids Res. 2013, 41, 8995–9005. [CrossRef] [PubMed] Metabolites 2021, 11, 572 16 of 21
- Szent-Gyorgyi, A. The living state and cancer. Proc. Natl. Acad. Sci. USA 1977, 74, 2844–2847. [CrossRef] [PubMed]
- Cairns, J. The origin of human cancers. Nature 1981, 289, 353–357. [CrossRef]
- Mukherjee, S. The Emperor of All Maladies: A Biography of Cancer (pages 285, 303, 333, 342); Scribner: New York, NK, USA, 2010; 579p. 65. Hu, Y.; Lu, W.; Chen, G.; Wang, P.; Chen, Z.; Zhou, Y.; Ogasawara, M.; Trachootham, D.; Feng, L.; Pelicano, H.; et al. K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012, 22, 399–412. [CrossRef] 66. Seyfried, T.N. Genes, respiration, viruses, and cancer. Chapter 9. In Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer; John Wiley & Sons: Hoboken, NJ, USA, 2012; pp. 145–176. 67. Seyfried, T.N. Respiratory dysfunction in cancer cells. Chapter 5. In Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer; John Wiley & Sons: Hoboken, NJ, USA, 2012; pp. 73–105. 68. Seyfried, T.N.; Shelton, L.M. Cancer as a metabolic disease. Nutr. Metab. 2010, 7, 7. [CrossRef] [PubMed] 69. Seyfried, T.N. Respiratory insufficiency, the retrograde response, and the origin of cancer. Chapter 10. In Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer; John Wiley & Sons: Hoboken, NJ, USA, 2012; pp. 177–194. 70. Seyfried, T.N. Mitochondrial respiratory dysfunction and the extrachromosomal origin of cancer. Chapter 14. In Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer; John Wiley & Sons: Hoboken, NJ, USA, 2012; pp. 253–259. 71. Zhu, Y.; Dean, A.E.; Horikoshi, N.; Heer, C.; Spitz, D.R.; Gius, D. Emerging evidence for targeting mitochondrial metabolic dysfunction in cancer therapy. J. Clin. Investig. 2018, 128, 3682–3691. [CrossRef] 72. Seyfried, T.N. Abnormalities in growth control, telomerase activity, apoptosis, and angiogenesis linked to mitochondrial dysfunction. Chapter 12. In Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer; John Wiley & Sons: Hoboken, NJ, USA, 2012; pp. 207–213. 73. Seoane, M.; Mosquera-Miguel, A.; Gonzalez, T.; Fraga, M.; Salas, A.; Costoya, J.A. The Mitochondrial Genome Is a “Genetic Sanctuary” during the Oncogenic Process. PLoS ONE 2011, 6, e23327. [CrossRef] 74. Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [CrossRef] 75. Srinivasan, S.; Guha, M.; Dong, D.W.; Whelan, K.A.; Ruthel, G.; Uchikado, Y.; Natsugoe, S.; Nakagawa, H.; Avadhani, N.G. Disruption of cytochrome c oxidase function induces the Warburg effect and metabolic reprogramming. Oncogene 2016, 35, 1585–1595. [CrossRef] 76. Ryan, M.T.; Hoogenraad, N.J. Mitochondrial-nuclear communications. Annu. Rev. Biochem. 2007, 76, 701–722. [CrossRef] [PubMed] 77. Biswas, G.; Srinivasan, S.; Anandatheerthavarada, H.K.; Avadhani, N.G. Dioxin-mediated tumor progression through activation of mitochondria-to-nucleus stress signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 186–191. [CrossRef] 78. Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [CrossRef] [PubMed] 79. Dang, C.V.; Le, A.; Gao, P. MYC-Induced Cancer Cell Energy Metabolism and Therapeutic Opportunities. Clin. Cancer Res. 2009, 15, 6479–6483. [CrossRef] [PubMed] 80. Dang, C.V.; Semenza, G.L. Oncogenic alterations of metabolism. Trends Biochem. Sci. 1999, 24, 68–72. [CrossRef] 81. Yang, D.; Kim, J. Mitochondrial Retrograde Signalling and Metabolic Alterations in the Tumour Microenvironment. Cells 2019, 8, 275. [CrossRef] [PubMed] 82. Semenza, G.L. Hypoxia-inducible factors: Coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. EMBO J. 2017, 36, 252–259. [CrossRef] [PubMed] 83. Seyfried, T.N.; Arismendi-Morillo, G.; Mukherjee, P.; Chinopoulos, C. On the Origin of ATP Synthesis in Cancer. iScience 2020, 23, 101761. [CrossRef] [PubMed] 84. Chinopoulos, C. From Glucose to Lactate and Transiting Intermediates Through Mitochondria, Bypassing Pyruvate Kinase: Considerations for Cells Exhibiting Dimeric PKM2 or Otherwise Inhibited Kinase Activity. Front. Physiol. 2020, 11, 543564. [CrossRef] 85. Seyfried, T.N. Metastasis. Chapter 13. In Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer; John Wiley & Sons: Hoboken, NJ, USA, 2012; pp. 215–252. 86. Taeubner, J.; Wieczorek, D.; Yasin, L.; Brozou, T.; Borkhardt, A.; Kuhlen, M. Penetrance and Expressivity in Inherited Cancer Predisposing Syndromes. Trends Cancer 2018, 4, 718–728. [CrossRef] [PubMed] 87. Shiovitz, S.; Korde, L.A. Genetics of breast cancer: A topic in evolution. Ann. Oncol. 2015, 26, 1291–1299. [CrossRef] 88. Anglian Breast Cancer Study Group. Prevalence and penetrance of BRCA1 and BRCA2 mutations in a population-based series of breast cancer cases. Anglian Breast Cancer Study Group. Br. J. Cancer 2000, 83, 1301–1308. [CrossRef] 89. Yuan, Y.; Ju, Y.S.; Kim, Y.; Li, J.; Wang, Y.; Yoon, C.J.; Yang, Y.; Martincorena, I.; Creighton, C.J.; Weinstein, J.N.; et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat. Genet. 2020, 52, 342–352. [CrossRef] 90. Kiebish, M.A.; Seyfried, T.N. Absence of pathogenic mitochondrial DNA mutations in mouse brain tumors. BMC Cancer 2005, 5, 102. [CrossRef] 91. Cruz-Bermúdez, A.; Vallejo, C.G.; Vicente-Blanco, R.J.; Gallardo, M.E.; Fernández-Moreno, M.Á.; Quintanilla, M.; Garesse, R. Enhanced tumorigenicity by mitochondrial DNA mild mutations. Oncotarget 2015, 6, 13628–13643. [CrossRef] Metabolites 2021, 11, 572 17 of 21 92. Elliott, R.L.; Jiang, X.P.; Head, J.F. Mitochondria organelle transplantation: Introduction of normal epithelial mitochondria into human cancer cells inhibits proliferation and increases drug sensitivity. Breast Cancer Res. Treat. 2012, 136, 347–354. [CrossRef] 93. Hall, A.; Meyle, K.D.; Lange, M.K.; Klima, M.; Sanderhoff, M.; Dahl, C.; Abildgaard, C.; Thorup, K.; Moghimi, S.M.; Jensen, P.B.; et al. Dysfunctional oxidative phosphorylation makes malignant melanoma cells addicted to glycolysis driven by the V600EBRAF oncogene. Oncotarget 2013, 4, 584–599. [CrossRef] 94. Seyfried, T.N. Energetics of normal cells and cancer cells. Chapter 4. In Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer; John Wiley & Sons: Hoboken, NJ, USA, 2012; pp. 47–72. 95. Seyfried, T.N. Perspectives on brain tumor formation involving macrophages, glia, and neural stem cells. Perspect. Biol. Med. 2001, 44, 263–282. [CrossRef] 96. McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; Zheng, L.; Gardet, A.; Tong, Z.; Jany, S.S.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238242. 97. Marsh, J.; Mukherjee, P.; Seyfried, T.N. Akt-dependent proapoptotic effects of dietary restriction on late-stage management of a phosphatase and tensin homologue/tuberous sclerosis complex 2-deficient mouse astrocytoma. Clin. Cancer Res. 2008, 14, 7751–7762. [CrossRef] [PubMed] 98. Kwong, J.Q.; Henning, M.S.; Starkov, A.A.; Manfredi, G. The mitochondrial respiratory chain is a modulator of apoptosis. J. Cell Biol. 2007, 179, 1163–1177. [CrossRef] 99. Laberge, G.S.; Duvall, E.; Haedicke, K.; Pawelek, J. Leukocyte–Cancer Cell Fusion-Genesis of a Deadly Journey. Cells 2019, 8, 170. [CrossRef] [PubMed] 100. Huysentruyt, L.C.; Mukherjee, P.; Banerjee, D.; Shelton, L.M.; Seyfried, T.N. Metastatic cancer cells with macrophage properties: Evidence from a new murine tumor model. Int. J. Cancer 2008, 123, 73–84. [CrossRef] [PubMed] 101. Huysentruyt, L.C.; Akgoc, Z.; Seyfried, T.N. Hypothesis: Are neoplastic macrophages/microglia present in glioblastoma multiforme? ASN Neuro 2011, 3, AN20110011. [CrossRef] 102. Garvin, S.; Patil, E.V.; Arnesson, L.G.; Oda, H.; Hedayati, E.; Lindström, A.; Shabo, I. Differences in intra-tumoral macrophage infiltration and radiotherapy response among intrinsic subtypes in pT1-T2 breast cancers treated with breast-conserving surgery. Virchows Arch. 2019, 475, 151–162. [CrossRef] 103. Powell, A.E.; Anderson, E.C.; Davies, P.S.; Silk, A.D.; Pelz, C.; Impey, S.; Wong, M.H. Fusion between Intestinal epithelial cells and macrophages in a cancer context results in nuclear reprogramming. Cancer Res. 2011, 71, 1497–1505. [CrossRef] 104. Shabo, I.; Midtbö, K.; Andersson, H.; Åkerlund, E.; Olsson, H.; Wegman, P.; Gunnarsson, C.; Lindström, A. Macrophage traits in cancer cells are induced by macrophage-cancer cell fusion and cannot be explained by cellular interaction. BMC Cancer 2015, 15, 922. [CrossRef] [PubMed] 105. Pawelek, J.M.; Chakraborty, A.K. Fusion of tumour cells with bone marrow-derived cells: A unifying explanation for metastasis. Nat. Rev. Cancer 2008, 8, 377–386. [CrossRef] [PubMed] 106. Ruff, M.R.; Pert, C.B. Origin of human small cell lung cancer. Science 1985, 229, 680. [CrossRef] [PubMed] 107. Wang, H.F.; Xiang, W.; Xue, B.Z.; Wang, Y.H.; Yi, D.Y.; Jiang, X.B.; Zhao, H.Y.; Fu, P. Cell fusion in cancer hallmarks: Current research status and future indications. Oncol. Lett. 2021, 22, 530. [CrossRef] 108. Schafer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell Biol. 2008, 9, 628–638. [CrossRef] [PubMed] 109. Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. 110. Ribatti, D.; Tamma, R. A revisited concept. Tumors: Wounds that do not heal. Crit. Rev. Oncol. Hematol. 2018, 128, 65–69. [CrossRef] 111. Huysentruyt, L.C.; Seyfried, T.N. Perspectives on the mesenchymal origin of metastatic cancer. Cancer Metastasis Rev. 2010, 29, 695–707. [CrossRef] 112. Jennings, J.F.; Oates, C.M. Studies on the nonspecific depression of the immune response. J. Exp. Med. 1967, 126, 557–564. [CrossRef] 113. Pryjma, J.; Ptak, W.; Szybinski, Z.; Sarnowicz, K. The macrophage in the antibody-mediated suppression of the humoral response. Int. Arch. Allergy Appl. Immunol. 1972, 43, 107–117. [CrossRef] 114. Duelli, D.; Lazebnik, Y. Cell fusion: A hidden enemy? Cancer Cell 2003, 3, 445–448. [CrossRef] 115. Seyfried, T.N.; Shelton, L.; Arismendi-Morillo, G.; Kalamian, M.; Elsakka, A.; Maroon, J.; Mukherjee, P. Provocative Question: Should Ketogenic Metabolic Therapy Become the Standard of Care for Glioblastoma? Neurochem. Res. 2019, 44, 2392–2404. [CrossRef] [PubMed] 116. Davies, P.S.; Powell, A.E.; Swain, J.R.; Wong, M.H. Inflammation and proliferation act together to mediate intestinal cell fusion. PLoS ONE 2009, 4, e6530. [CrossRef] [PubMed] 117. Cruzat, V.; Macedo Rogero, M.; Noel Keane, K.; Curi, R.; Newsholme, P. Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients 2018, 10, 1564. [CrossRef] 118. Newsholme, P. Why is L-glutamine metabolism important to cells of the immune system in health, postinjury, surgery or infection? J. Nutr. 2001, 131 (Suppl. S9), 2515S–2522S; discussion 23S–24S. [CrossRef] 119. Newsholme, P.; Curi, R.; Gordon, S.; Newsholme, E.A. Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochem. J. 1986, 239, 121–125. [CrossRef] Metabolites 2021, 11, 572 18 of 21 120. Flores, R.E.; Brown, A.K.; Taus, L.; Khoury, J.; Glover, F.; Kami, K.; Sarangarajan, R.; Walshe, T.E.; Narain, N.R.; Kiebish, M.A.; et al. Mycoplasma infection and hypoxia initiate succinate accumulation and release in the VM-M3 cancer cells. Biochim. Biophys. Acta 2018, 1859, 975–983. [CrossRef] 121. Shelton, L.M.; Huysentruyt, L.C.; Seyfried, T.N. Glutamine targeting inhibits systemic metastasis in the VM-M3 murine tumor model. Int. J. Cancer 2010, 127, 2478–2485. [CrossRef] 122. van den Heuvel, A.P.; Jing, J.; Wooster, R.F.; Bachman, K.E. Analysis of glutamine dependency in non-small cell lung cancer: GLS1 splice variant GAC is essential for cancer cell growth. Cancer Biol. Ther. 2012, 13, 1185–1194. [CrossRef] 123. Mukherjee, P.; Augur, Z.M.; Li, M.; Hill, C.; Greenwood, B.; Domin, M.A.; Kondakci, G.; Narain, N.R.; Kiebish, M.A.; Bronson, R.T.; et al. Therapeutic benefit of combining calorie-restricted ketogenic diet and glutamine targeting in late-stage experimental glioblastoma. Commun. Biol. 2019, 2, 200. [CrossRef] 124. Ladanie, A.; Schmitt, A.M.; Speich, B.; Naudet, F.; Agarwal, A.; Pereira, T.V.; Sclafani, F.; Herbrand, A.K.; Briel, M.; Martin-Liberal, J.; et al. Clinical Trial Evidence Supporting US Food and Drug Administration Approval of Novel Cancer Therapies between 2000 and 2016. JAMA Netw. Open 2020, 3, e2024406. [CrossRef] 125. Ferrara, R.; Mezquita, L.; Texier, M.; Lahmar, J.; Audigier-Valette, C.; Tessonnier, L.; Mazieres, J.; Zalcman, G.; Brosseau, S.; Le Moulec, S.; et al. Hyperprogressive Disease in Patients with Advanced Non-Small Cell Lung Cancer Treated with PD-1/PD-L1 Inhibitors or with Single-Agent Chemotherapy. JAMA Oncol. 2018, 4, 1543–1552. [CrossRef] [PubMed] 126. Pelicano, H.; Xu, R.H.; Du, M.; Feng, L.; Sasaki, R.; Carew, J.S.; Hu, Y.; Ramdas, L.; Hu, L.; Keating, M.J.; et al. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J. Cell Biol. 2006, 175, 913–923. [CrossRef] [PubMed] 127. Xu, R.H.; Pelicano, H.; Zhou, Y.; Carew, J.S.; Feng, L.; Bhalla, K.N.; Keating, M.J.; Huang, P. Inhibition of glycolysis in cancer cells: A novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005, 65, 613–621. [PubMed] 128. Pawelek, J.M. Fusion of bone marrow-derived cells with cancer cells: Metastasis as a secondary disease in cancer. Chin. J. Cancer 2014, 33, 133–139. [CrossRef] [PubMed] 129. Pawelek, J.M.; Chakraborty, A.K. The cancer cell–leukocyte fusion theory of metastasis. Adv. Cancer Res. 2008, 101, 397–444. 130. Munzarova, M.; Kovarik, J. Is cancer a macrophage-mediated autoaggressive disease? Lancet 1987, 1, 952–954. [CrossRef] 131. Munzarova, M.; Lauerova, L.; Capkova, J. Are advanced malignant melanoma cells hybrids between melanocytes and macrophages? Melanoma Res. 1992, 2, 127–129. [CrossRef] 132. Munzarova, M.; Lauerova, L.; Kovarik, J.; Rejthar, A.; Brezina, V.; Kellnerova, R.; Kovarik, A. Fusion-induced malignancy? A preliminary study (a challenge to today’s common wisdom). Neoplasma 1992, 39, 79–86. 133. Whalen, G.F. Solid tumours and wounds: Transformed cells misunderstood as injured tissue? Lancet 1990, 336, 1489–1492. [CrossRef] 134. Weinberg, R.A. The Biology of Cancer; Garland Science: New York, NY, USA, 2007; 796p. 135. Lobikin, M.; Chernet, B.; Lobo, D.; Levin, M. Resting potential, oncogene-induced tumorigenesis, and metastasis: The bioelectric basis of cancer in vivo. Phys. Biol. 2012, 9, 065002. [CrossRef] [PubMed] 136. Chernet, B.; Levin, M. Endogenous Voltage Potentials and the Microenvironment: Bioelectric Signals that Reveal, Induce and Normalize Cancer. J. Clin. Exp. Oncol. 2013. [CrossRef] 137. Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [CrossRef] [PubMed] 138. Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [CrossRef] 139. Frost, M.T.; Wang, Q.; Moncada, S.; Singer, M. Hypoxia accelerates nitric oxide-dependent inhibition of mitochondrial complex I in activated macrophages. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R394–R400. [CrossRef] 140. Erusalimsky, J.D.; Moncada, S. Nitric oxide and mitochondrial signaling: From physiology to pathophysiology. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2524–2531. [CrossRef] 141. Li, N.; Sioutas, C.; Cho, A.; Schmitz, D.; Misra, C.; Sempf, J.; Wang, M.; Oberley, T.; Froines, J.; Nel, A. Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ. Health Perspect. 2003, 111, 455–460. [CrossRef] 142. Bierie, B.; Moses, H.L. Tumour microenvironment: TGFbeta: The molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 2006, 6, 506–520. [CrossRef] 143. Bierie, B.; Moses, H.L. TGF-beta and cancer. Cytokine Growth Factor Rev. 2006, 17, 29–40. [CrossRef] 144. Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, 320–329. [CrossRef] 145. Yoon, Y.S.; Lee, J.H.; Hwang, S.C.; Choi, K.S.; Yoon, G. TGF beta1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene 2005, 24, 1895–1903. [CrossRef] 146. Ortega, A.D.; Sanchez-Arago, M.; Giner-Sanchez, D.; Sanchez-Cenizo, L.; Willers, I.; Cuezva, J.M. Glucose avidity of carcinomas. Cancer Lett. 2009, 276, 125–135. [CrossRef] [PubMed] 147. Moiseeva, O.; Bourdeau, V.; Roux, A.; Deschenes-Simard, X.; Ferbeyre, G. Mitochondrial dysfunction contributes to oncogene[1]induced senescence. Mol. Cell. Biol. 2009, 29, 4495–4507. [CrossRef] [PubMed] 148. Sonnenschein, C.; Soto, A.M. The Society of Cells: Cancer and the Control of Cell Proliferation; Springer: New York, NY, USA, 1999; 154p. Metabolites 2021, 11, 572 19 of 21 149. Sonnenschein, C.; Soto, A.M. Theories of carcinogenesis: An emerging perspective. Semin. Cancer Biol. 2008, 18, 372–377. [CrossRef] 150. Sonnenschein, C.; Soto, A.M. An Integrative Approach toward Biology, Organisms, and Cancer. Methods Mol. Biol. 2018, 1702, 15–26. 151. Soto, A.M.; Sonnenschein, C. Is systems biology a promising approach to resolve controversies in cancer research? Cancer Cell Int. 2012, 12, 12. [CrossRef] [PubMed] 152. Zhang, L.; Bell, R.J.; Kiebish, M.A.; Seyfried, T.N.; Han, X.; Gross, R.W.; Chuang, J.H. A mathematical model for the determination of steady-state cardiolipin remodeling mechanisms using lipidomic data. PLoS ONE 2011, 6, e21170. [CrossRef] 153. Kiebish, M.A.; Han, X.; Cheng, H.; Chuang, J.H.; Seyfried, T.N. Cardiolipin and electron transport chain abnormalities in mouse brain tumor mitochondria: Lipidomic evidence supporting the Warburg theory of cancer. J. Lipid Res. 2008, 49, 2545–2556. [CrossRef] [PubMed] 154. Arismendi-Morillo, G.; Castellano-Ramirez, A.; Seyfried, T.N. Ultrastructural characterization of the Mitochondria-associated membranes abnormalities in human astrocytomas: Functional and therapeutics implications. Ultrastruct Pathol. 2017, 41, 234–244. [CrossRef] 155. Simoes, I.C.; Morciano, G.; Lebiedzinska-Arciszewska, M.; Aguiari, G.; Pinton, P.; Potes, Y.; Wieckowski, M.R. The mystery of mitochondria-ER contact sites in physiology and pathology: A cancer perspective. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165834. [CrossRef] 156. Lehninger, A.L. The Mitochondrion: Molecular Basis of Structure and Function; W.A. Benjamin, Inc.: New York, NY, USA, 1964; 263p. 157. Stroud, D.A.; Ryan, M.T. Mitochondria: Organization of respiratory chain complexes becomes cristae-lized. Curr. Biol. 2013, 23, R969–R971. [CrossRef] [PubMed] 158. Hackenbrock, C.R. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. II. Electron transport-linked ultrastructural transformations in mitochondria. J. Cell Biol. 1968, 37, 345–369. [CrossRef] 159. Putignani, L.; Raffa, S.; Pescosolido, R.; Rizza, T.; Del Chierico, F.; Leone, L.; Aimati, L.; Signore, F.; Carrozzo, R.; Callea, F.; et al. Preliminary evidences on mitochondrial injury and impaired oxidative metabolism in breast cancer. Mitochondrion 2012, 12, 363–369. [CrossRef] [PubMed] 160. Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273. [CrossRef] [PubMed] 161. Warburg, O. The Metabolism of Tumours; Richard R. Smith Inc.: New York, NY, USA, 1931; 327p. 162. Ta, N.L.; Seyfried, T.N. Influence of Serum and Hypoxia on Incorporation of [(14)C]-D-Glucose or [(14)C]-L-Glutamine into Lipids and Lactate in Murine Glioblastoma Cells. Lipids 2015, 50, 1167–1184. [CrossRef] 163. Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [CrossRef] 164. Barron, E.S. The Catalytic Effect of Methylene Blue on the Oxygen Consumption of Tumors and Normal Tissues. J. Exp. Med. 1930, 52, 447–456. [CrossRef] 165. Renner, C.; Asperger, A.; Seyffarth, A.; Meixensberger, J.; Gebhardt, R.; Gaunitz, F. Carnosine inhibits ATP production in cells from malignant glioma. Neurol Res. 2010, 32, 101–105. [CrossRef] 166. Ceruti, S.; Mazzola, A.; Abbracchio, M.P. Resistance of human astrocytoma cells to apoptosis induced by mitochondria-damaging agents: Possible implications for anticancer therapy. J. Pharmacol. Exp. Ther. 2005, 314, 825–837. [CrossRef] 167. Seyfried, T.N. Is mitochondrial glutamine fermentation a missing link in the metabolic theory of cancer? Chapter 8. In Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer; John Wiley & Sons: Hoboken, NJ, USA, 2012; pp. 133–144. 168. Yu, M.; Chen, S.; Hong, W.; Gu, Y.; Huang, B.; Lin, Y.; Zhou, Y.; Jin, H.; Deng, Y.; Tu, L.; et al. Prognostic role of glycolysis for cancer outcome: Evidence from 86 studies. J. Cancer Res. Clin. Oncol. 2019, 145, 967–999. [CrossRef] 169. Burk, D.; Woods, M.; Hunter, J. On the significance of glucolysis for cancer growth, with special reference to Morris rat hepatomas. J. Natl. Cancer Inst. 1967, 38, 839–863. 170. Israelsen, W.J.; Dayton, T.L.; Davidson, S.M.; Fiske, B.P.; Hosios, A.M.; Bellinger, G.; Li, J.; Yu, Y.; Sasaki, M.; Horner, J.W.; et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 2013, 155, 397–409. [CrossRef] 171. Boros, L.G.; Lee, P.W.; Brandes, J.L.; Cascante, M.; Muscarella, P.; Schirmer, W.J.; Melvin, W.S.; Ellison, E.C. Nonoxidative pentose phosphate pathways and their direct role in ribose synthesis in tumors: Is cancer a disease of cellular glucose metabolism? Med. Hypotheses 1998, 50, 55–59. [CrossRef] 172. Vander Heiden, M.G.; Locasale, J.W.; Swanson, K.D.; Sharfi, H.; Heffron, G.J.; Amador-Noguez, D.; Christofk, H.R.; Wagner, G.; Rabinowitz, J.D.; Asara, J.M.; et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science 2010, 329, 1492–1499. [CrossRef] 173. Kathagen, A.; Schulte, A.; Balcke, G.; Phillips, H.S.; Martens, T.; Matschke, J.; Günther, H.S.; Soriano, R.; Modrusan, Z.; Sandmann, T.; et al. Hypoxia and oxygenation induce a metabolic switch between pentose phosphate pathway and glycolysis in glioma stem-like cells. Acta Neuropathol. 2013, 126, 763–780. [CrossRef] [PubMed] 174. Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [CrossRef] [PubMed] 175. Mazat, J.P. One-carbon metabolism in cancer cells: A critical review based on a core model of central metabolism. Biochem. Soc. Trans. 2021, 49, 1–15. [CrossRef] [PubMed] Metabolites 2021, 11, 572 20 of 21 176. Vozza, A.; Parisi, G.; De Leonardis, F.; Lasorsa, F.M.; Castegna, A.; Amorese, D.; Marmo, R.; Calcagnile, V.M.; Palmieri, L.; Ricquier, D.; et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, 960–965. [CrossRef] [PubMed] 177. Samudio, I.; Fiegl, M.; Andreeff, M. Mitochondrial uncoupling and the Warburg effect: Molecular basis for the reprogramming of cancer cell metabolism. Cancer Res. 2009, 69, 2163–2166. [CrossRef] 178. Giudetti, A.M.; De Domenico, S.; Ragusa, A.; Lunetti, P.; Gaballo, A.; Franck, J.; Simeone, P.; Nicolardi, G.; De Nuccio, F.; Santino, A.; et al. A specific lipid metabolic profile is associated with the epithelial mesenchymal transition program. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 344–357. [CrossRef] 179. Andersen, K.F.; Divilov, V.; Sevak, K.; Koziorowski, J.; Lewis, J.S.; Pillarsetty, N. Influence of free fatty acids on glucose uptake in prostate cancer cells. Nucl. Med. Biol. 2014, 41, 254–258. [CrossRef] 180. Wagner, B.A.; Venkataraman, S.; Buettner, G.R. The rate of oxygen utilization by cells. Free Radic. Biol. Med. 2011, 51, 700–712. [CrossRef] 181. Hochachka, P.W.; Somero, G.N. Biochemical Adaptation: Mechanism and Process in Physiological Evolution; Oxford Press: New York, NY, USA, 2002; 466p. 182. Chinopoulos, C.; Seyfried, T.N. Mitochondrial Substrate-Level Phosphorylation as Energy Source for Glioblastoma: Review and Hypothesis. ASN Neuro 2018, 10, 1759091418818261. [CrossRef] 183. Chinopoulos, C. Acute sources of mitochondrial NAD(+) during respiratory chain dysfunction. Exp. Neurol. 2020, 327, 113218. [CrossRef] 184. Chinopoulos, C. Mitochondrial consumption of cytosolic ATP: Not so fast. FEBS Lett. 2011, 585, 1255–1259. [CrossRef] [PubMed] 185. Chinopoulos, C. The “B space” of mitochondrial phosphorylation. J. Neurosci. Res. 2011, 89, 1897–1904. [CrossRef] [PubMed] 186. Ahluwalia, G.S.; Grem, J.L.; Hao, Z.; Cooney, D.A. Metabolism and action of amino acid analog anti-cancer agents. Pharmacol. Ther. 1990, 46, 243–271. [CrossRef] 187. Young, V.R.; Ajami, A.M. Glutamine: The emperor or his clothes? J. Nutr. 2001, 131 (Suppl. S9), 2449S–2459S; discussion 86S–87S. [CrossRef] [PubMed] 188. Panosyan, E.H.; Lin, H.J.; Koster, J.; Lasky, J.L., 3rd. In search of druggable targets for GBM amino acid metabolism. BMC Cancer 2017, 17, 162. [CrossRef] [PubMed] 189. Knott, S.R.; Wagenblast, E.; Khan, S.; Kim, S.Y.; Soto, M.; Wagner, M.; Turgeon, M.O.; Fish, L.; Erard, N.; Gable, A.L.; et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 2018, 554, 378–381. [CrossRef] 190. DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [CrossRef] [PubMed] 191. DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [CrossRef] [PubMed] 192. Portais, J.C.; Voisin, P.; Merle, M.; Canioni, P. Glucose and glutamine metabolism in C6 glioma cells studied by carbon 13 NMR. Biochimie 1996, 78, 155–164. [CrossRef] 193. Reitzer, L.J.; Wice, B.M.; Kennell, D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J. Biol. Chem. 1979, 254, 2669–2676. [CrossRef] 194. Scott, D.A.; Richardson, A.D.; Filipp, F.V.; Knutzen, C.A.; Chiang, G.G.; Ze’ev, A.R.; Osterman, A.L.; Smith, J.W. Comparative metabolic flux profiling of melanoma cell lines: Beyond the Warburg effect. J. Biol. Chem. 2011, 286, 42626–42634. [CrossRef] 195. Gillies, R.J.; Robey, I.; Gatenby, R.A. Causes and consequences of increased glucose metabolism of cancers. J. Nucl. Med. 2008, 49 (Suppl. 2), 24S–42S. [CrossRef] 196. Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [CrossRef] [PubMed] 197. Sanadi, D.R.; Gibson, D.M.; Ayengar, P.; Jacob, M. Alpha-ketoglutaric dehydrogenase. V. Guanosine diphosphate in coupled phosphorylation. J. Biol. Chem. 1956, 218, 505–520. [CrossRef] 198. Kaufman, S.; Gilvarg, C.; Cori, O.; Ochoa, S. Enzymatic oxidation of alpha-ketoglutarate and coupled phosphorylation. J. Biol. Chem. 1953, 203, 869–888. [CrossRef] 199. Ottaway, J.H.; McClellan, J.A.; Saunderson, C.L. Succinic thiokinase and metabolic control. Int. J. Biochem. 1981, 13, 401–410. [CrossRef] 200. Hunter, F.E., Jr.; Hixon, W.S. Phosphorylation coupled with the oxidation of alpha-ketoglutaric acid. J. Biol. Chem. 1949, 181, 73–79. [CrossRef] 201. Chen, Q.; Kirk, K.; Shurubor, Y.I.; Zhao, D.; Arreguin, A.J.; Shahi, I.; Valsecchi, F.; Primiano, G.; Calder, E.L.; Carelli, V.; et al. Rewiring of Glutamine Metabolism Is a Bioenergetic Adaptation of Human Cells with Mitochondrial DNA Mutations. Cell Metab. 2018, 27, 1007–1025.e5. [CrossRef] 202. Auger, C.; Vinaik, R.; Appanna, V.D.; Jeschke, M.G. Beyond mitochondria: Alternative energy-producing pathways from all strata of life. Metabolism 2021, 118, 154733. [CrossRef] 203. Lin, S.R.; Wen, Y.C.; Yeh, H.L.; Jiang, K.C.; Chen, W.H.; Mokgautsi, N.; Huang, J.; Chen, W.Y.; Liu, Y.N. EGFR-upregulated LIFR promotes SUCLG2-dependent castration resistance and neuroendocrine differentiation of prostate cancer. Oncogene 2020, 39, 6757–6775. [CrossRef] Metabolites 2021, 11, 572 21 of 21 204. Gao, C.; Shen, Y.; Jin, F.; Miao, Y.; Qiu, X. Cancer Stem Cells in Small Cell Lung Cancer Cell Line H446: Higher Dependency on Oxidative Phosphorylation and Mitochondrial Substrate-Level Phosphorylation than Non-Stem Cancer Cells. PLoS ONE 2016, 11, e0154576. [CrossRef] 205. Kiebish, M.A.; Han, X.; Cheng, H.; Seyfried, T.N. In vitro growth environment produces lipidomic and electron transport chain abnormalities in mitochondria from non-tumorigenic astrocytes and brain tumours. ASN Neuro 2009, 1, e00011. [CrossRef] 206. Dufort, F.J.; Gumina, M.R.; Ta, N.L.; Tao, Y.; Heyse, S.A.; Scott, D.A.; Richardson, A.D.; Seyfried, T.N.; Chiles, T.C. Glucose[1]dependent de novo lipogenesis in B lymphocytes: A requirement for atp-citrate lyase in lipopolysaccharide-induced differentia[1]tion. J. Biol. Chem. 2014, 289, 7011–7024. [CrossRef] [PubMed] 207. Klarquist, J.; Chitrakar, A.; Pennock, N.D.; Kilgore, A.M.; Blain, T.; Zheng, C.; Danhorn, T.; Walton, K.; Jiang, L.; Sun, J.; et al. Clonal expansion of vaccine-elicited T cells is independent of aerobic glycolysis. Sci. Immunol. 2018, 3, eaas9822. [CrossRef] [PubMed] 208. Hague, A.; Singh, B.; Paraskeva, C. Butyrate acts as a survival factor for colonic epithelial cells: Further fuel for the in vivo versus in vitro debate. Gastroenterology 1997, 112, 1036–1040. [CrossRef] [PubMed] 209. Simek, J.; Sedlacek, J. Effect of glucose administered in vivo or in vitro on the respiratory quotient of rat liver tissue after partial hepatectomy. Nature 1965, 207, 761–762. [CrossRef] [PubMed] 210. Thevananther, S. Adipose to the rescue: Peripheral fat fuels liver regeneration. Hepatology 2010, 52, 1875–1876. [CrossRef] 211. O’Connor, R.J. The effect on cell division of inhibiting aerobic glycolysis. Br. J. Exp. Pathol. 1950, 31, 449–453. [PubMed] 212. Paul, D. Cancer as a form of life: Musings of the cancer and evolution symposium. Prog. Biophys. Mol. Biol. 2021. [CrossRef] [PubMed] 213. Seyfried, T.N.; Shivane, A.G.; Kalamian, M.; Maroon, J.C.; Mukherjee, P.; Zuccoli, G. Ketogenic Metabolic Therapy, Without Chemo or Radiation, for the Long-Term Management of IDH1-Mutant Glioblastoma: An 80-Month Follow-Up Case Report. Front. Nutr. 2021, 8, 682243. [CrossRef] 214. Zhang, E.; Xu, H. A new insight in chimeric antigen receptor-engineered T cells for cancer immunotherapy. J. Hematol. Oncol. 2017, 10, 1. [CrossRef] [PubMed] 215. Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2- hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [CrossRef] 216. Zhang, K.; Xu, P.; Sowers, J.L.; Machuca, D.F.; Mirfattah, B.; Herring, J.; Tang, H.; Chen, Y.; Tian, B.; Brasier, A.R.; et al. Proteome Analysis of Hypoxic Glioblastoma Cells Reveals Sequential Metabolic Adaptation of One-Carbon Metabolic Pathways. Mol. Cell. Proteom. 2017, 16, 1906–1921. [CrossRef] [PubMed] 217. Chesnelong, C.; Chaumeil, M.M.; Blough, M.D.; Al-Najjar, M.; Stechishin, O.D.; Chan, J.A.; Pieper, R.O.; Ronen, S.M.; Weiss, S.; Luchman, H.A.; et al. Lactate dehydrogenase A silencing in IDH mutant gliomas. Neuro Oncol. 2014, 16, 686–695. [CrossRef] 218. Arens, N.C.; West, I.D. Press-pulse: A general theory of mass extinction? Paleobiology 2008, 34, 456–471. [CrossRef] 219. Potts, R. Complexity of Adaptibility in Human Evolution. In Probing Human Origins; Goodman, M., Moffat, A.S., Eds.; American Academy of Arts & Sciences: Cambridge, MA, USA, 2002; pp. 33–57. 220. Potts, R. Humanity’s Descent: The Consequences of Ecological Instability; William Morrow & Co., Inc.: New York, NY, USA, 1996; 325p. 221. Seyfried, T.N. Metabolic management of cancer. Chapter 17. In Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer; John Wiley & Sons: Hoboken, NJ, USA, 2012; pp. 291–354. 222. Drenick, E.J.; Alvarez, L.C.; Tamasi, G.C.; Brickman, A.S. Resistance to symptomatic insulin reactions after fasting. J. Clin. Investig. 1972, 51, 2757–2762. [CrossRef] 223. LaManna, J.C.; Salem, N.; Puchowicz, M.; Erokwu, B.; Koppaka, S.; Flask, C.; Lee, Z. Ketones suppress brain glucose consumption. Adv. Exp. Med. Biol. 2009, 645, 301–306. 224. VanItallie, T.B.; Nufert, T.H. Ketones: Metabolism’s ugly duckling. Nutr. Rev. 2003, 61, 327–341. [CrossRef] 225. Mukherjee, P.; Mulrooney, T.J.; Marsh, J.; Blair, D.; Chiles, T.C.; Seyfried, T.N. Differential effects of energy stress on AMPK phosphorylation and apoptosis in experimental brain tumor and normal brain. Mol. Cancer 2008, 7, 37. [CrossRef] 226. Mulrooney, T.J.; Marsh, J.; Urits, I.; Seyfried, T.N.; Mukherjee, P. Influence of Caloric Restriction on Constitutive Expression of NF-kappaB in an Experimental Mouse Astrocytoma. PLoS ONE 2011, 6, e18085. [CrossRef] [PubMed] 227. Seyfried, T.N.; Mukherjee, P. Targeting energy metabolism in brain cancer: Review and hypothesis. Nutr. Metab. 2005, 2, 30. [CrossRef] [PubMed] 228. Wagle, S.R.; Morris, H.P.; Weber, G. Comparative Biochemistry of Hepatomas. V. Studies on Amino Acid Incorporation in Liver Tumors of Different Growth Rates. Cancer Res. 1963, 23, 1003–1007. 229. Burk, D.; Behrens, O.K.; Sugiura, K. Metabolism of butter yellow rat liver cancers. Cancer Res. 1941, 1, 733–734. 230. Veech, R.L. The therapeutic implications of ketone bodies: The effects of ketone bodies in pathological conditions: Ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot Essent Fat. Acids 2004, 70, 309–319. [CrossRef] 231. Veech, R.L.; Todd King, M.; Pawlosky, R.; Kashiwaya, Y.; Bradshaw, P.C.; Curtis, W. The “great” controlling nucleotide coenzymes. IUBMB Life 2019, 71, 565–579. [CrossRef